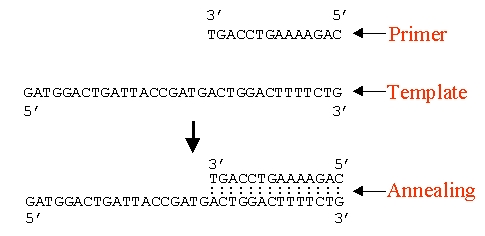
PCR (Polymerase Chain Reaction)
Polymerase Chain Reaction is widely held as one of the most important inventions of the 20th century in molecular biology. Small amounts of the genetic material can now be amplified to be able to a identify, manipulate DNA, detect infectious organisms, including the viruses that cause AIDS, hepatitis, tuberculosis, detect genetic variations, including mutations, in human genes and numerous other tasks.
PCR involves the following three steps: denaturation, annealing and extension. First, the genetic material is denatured, converting the double stranded DNA molecules to single strands. The primers are then annealed to the complementary regions of the single stranded molecules. In the third step, they are extended by the action of the DNA polymerase. All these steps are temperature sensitive and the common choice of temperatures is 94
oC, 60
oC and 70
oC respectively. Good primer design is essential for successful reactions. The important design considerations described below are a key to specific amplification with high yield. The preferred values indicated are built into all our products by default.
1. Primer Length: It is generally accepted that the optimal length of PCR primers is 18-22 bp. This length is long enough for adequate specificity, and short enough for primers to bind easily to the template at the annealing temperature.
2. Primer Melting Temperature: Primer Melting Temperature (Tm) by definition is the temperature at which one half of the DNA duplex will dissociate to become single stranded and indicates the duplex stability. Primers with melting temperatures in the range of 52-58 oC generally produce the best results. Primers with melting temperatures above 65oC have a tendency for secondary annealing. The GC content of the sequence gives a fair indication of the primer Tm. All our products calculate it using the nearest neighbor thermodynamic theory, accepted as a much superior method for estimating it, which is considered the most recent and best available.
Formula for primer Tm calculation:
Melting Temperature Tm(oK)={ΔH/ ΔS + R ln(C)}, Or Melting Temperature Tm(oC) = {ΔH/ ΔS + R ln(C)} - 273.15 where
ΔH (kcal/mole) : H is the Enthalpy. Enthalpy is the amount of heat energy possessed by substances. ΔH is the change in Enthalpy. In the above formula the ΔH is obtained by adding up all the di-nucleotide pairs enthalpy values of each nearest neighbor base pair.
ΔS (kcal/mole) : S is the amount of disorder a system exhibits is called entropy. ΔS is change in Entropy. Here it is obtained by adding up all the di-nucleotide pairs entropy values of each nearest neighbor base pair. An additional salt correction is added as the Nearest Neighbor parameters were obtained from DNA melting studies conducted in 1M Na+ buffer and this is the default condition used for all calculations.
ΔS (salt correction) = ΔS (1M NaCl )+ 0.368 x N x ln([Na+])
Where
N is the number of nucleotide pairs in the primer ( primer length -1).
[Na+] is salt equivalent in mM.
[Na+] calculation:
[Na+] = Monovalent ion concentration +4 x free Mg2+.
3.Primer annealing temperature : The primer melting temperature is the estimate of the DNA-DNA hybrid stability and critical in determining the annealing temperature. Too high Ta will produce insufficient primer-template hybridization resulting in low PCR product yield. Too low Ta may possibly lead to non-specific products caused by a high number of base pair mismatches,. Mismatch tolerance is found to have the strongest influence on PCR specificity.
Ta = 0.3 x Tm(primer) + 0.7 Tm (product) – 14.9
where,
Tm(primer) = Melting Temperature of the primers
Tm(product) = Melting temperature of the product
4. GC Content : The GC content (the number of G's and C's in the primer as a percentage of the total bases) of primer should be 40-60%.
5. GC Clamp : The presence of G or C bases within the lat five bases from the 3' end of primers (GC clamp) helps promote specific binding at the 3' end due to the stronger bonding of G and C bases. More than 3 G's or C's should be avoided in the last 5 bases at the 3' end of the primer.
6. Primer Secondary Structures : Presence of the primer secondary structures produced by intermolecular or intramolecular interactions can lead to poor or no yield of the product. They adversely affect primer template annealing and thus the amplification. They greatly reduce the availability of primers to the reaction.
i) Hairpins : It is formed by intramolecular interaction within the primer and should be avoided. Optimally a 3' end hairpin with a ΔG of -2 kcal/mol and an internal hairpin with a ΔG of -3 kcal/mol is tolerated generally.
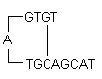
ΔG definition : The Gibbs Free Energy G is the measure of the amount of work that can be extracted from a process operating at a constant pressure. It is the measure of the spontaneity of the reaction. The stability of hairpin is commonly represented by its ΔG value, the energy required to break the secondary structure. Larger negative value for ΔG indicates stable, undesirable hairpins. Presence of hairpins at the 3' end most adversely affects the reaction.
ΔG = ΔH – TΔS
ii) Self Dimer : A primer self-dimer is formed by intermolecular interactions between the two (same sense) primers, where the primer is homologous to itself. Generally a large amount of primers are used in PCR compared to the amount of target gene. When primers form intermolecular dimers much more readily than hybridizing to target DNA, they reduce the product yield. Optimally a 3' end self dimer with a ΔG of -5 kcal/mol and an internal self dimer with a ΔG of -6 kcal/mol is tolerated generally.
iii) Cross Dimer : Primer cross dimers are formed by intermolecular interaction between sense and antisense primers, where they are homologous. Optimally a 3' end cross dimer with a ΔG of -5 kcal/mol and an internal cross dimer with a ΔG of -6 kcal/mol is tolerated generally.
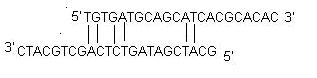
7. Repeats : A repeat is a di-nucleotide occurring many times consecutively and should be avoided because they can misprime. For example: ATATATAT. A maximum number of di-nucleotide repeats acceptable in an oligo is 4 di-nucleotides.
8. Runs : Primers with long runs of a single base should generally be avoided as they can misprime. For example, AGCGGGGGATGGGG has runs of base 'G' of value 5 and 4. A maximum number of runs accepted is 4bp.
9. 3' End Stability : It is the maximum ΔG value of the five bases from the 3' end. An unstable 3' end (less negative ΔG) will result in less false priming.
10. Avoid Template secondary structure : A single stranded Nucleic acid sequences is highly unstable and fold into conformations (secondary structures). The stability of these template secondary structures depends largely on their free energy and melting temperatures(Tm). Consideration of template secondary structures is important in designing primers, especially in qPCR. If primers are designed on a secondary structures which is stable even above the annealing temperatures, the primers are unable to bind to the template and the yield of PCR product is significantly affected. Hence, it is important to design primers in the regions of the templates that do not form stable secondary structures during the PCR reaction. Our products determine the secondary structures of the template and design primers avoiding them.
11. Avoid Cross homology : To improve specificity of the primers it is necessary to avoid regions of homology. Primers designed for a sequence must not amplify other genes in the mixture. Commonly, primers are designed and then BLASTed to test the specificity. Our products offer a better alternative. You can avoid regions of cross homology while designing primers. You can BLAST the templates against the appropriate non-redundant database and the software will interpret the results. It will identify regions significant cross homologies in each template and avoid them during primer search.
Parameters for Primer Pair Design:
1. Amplicon Length : The amplicon length is dictated by the experimental goals. For qPCR, the target length is closer to 100 bp and for standard PCR, it is near 500 bp. If you know the positions of each primer with respect to the template, the product is calculated as: Product length = (Position of antisense primer-Position of sense primer) + 1.
2. Product position : Primer can be located near the 5' end, the 3' end or any where within specified length. Generally, the sequence close to the 3' end is known with greater confidence and hence preferred most frequently.
3. Tm of Product : Melting Temperature (Tm) is the temperature at which one half of the DNA duplex will dissociate and become single stranded. The stability of the primer-template DNA duplex can be measured by the melting temperature (Tm).
4.Optimum Annealing temperature (Ta Opt): The formula of Rychlik is most respected. Our products use this formula to calculate it and thousands of our customers have reported good results using it for the annealing step of the PCR cycle. It usually results in good PCR product yield with minimum false product production.
Ta Opt = 0.3 x(Tm of primer) + 0.7 x(Tm of product) - 25
where
Tm of primer is the melting temperature of the less stable primer-template pair
Tm of product is the melting temperature of the PCR product.
5. Primer Pair Tm Mismatch Calculation : The two primers of a primer pair should have closely matched melting temperatures for maximizing PCR product yield. The difference of 5oC or more can lead no amplification.
Primer Design Using Software
A number of primer design tools are available that can assist in PCR primer design for new and experienced users alike. These tools may reduce the cost and time involved in experimentation by lowering the chances of failed experimentation.
Primer Premier follows all the guidelines specified for PCR primer design. Primer Premier can be used to design primers for single templates, alignments, degenerate primer design, restriction enzyme analysis. contig analysis and design of sequencing primers.
The guidelines for qPCR primer design vary slightly. Software such as AlleleID and Beacon Designer can design primers and oligonucleotide probes for complex detection assays such as multiplex assays, cross species primer design, species specific primer design and primer design to reduce the cost of experimentation.
PrimerPlex is a software that can design ASPE (Allele specific Primer Extension) primers and capture probes for multiplex SNP genotyping using suspension array systems such as Luminex xMAP® and BioRad Bioplex.
References :
1. “A critical review of PCR primer design algorithms and cross-hybridization case study” By F.John Burpo.
2. “Optimization of the annealing temperature for DNA amplification in vitro” By W.Rychlik, W.J.Spencer
and R.E.Rhoads.
3. “A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics” By John SantaLucia.
4. “A computer program for selection of oligonucleotide primers for polymerase chain reactions” Lowe T, Sharefkin J, Yang SQ, Dieffenbach CW.
5. “Optimization strategies for the polymerase chain reaction” Williams JF.Perkin-Elmer Corporation, Norwalk, CT 06859-0251.
6. “Algorithms and thermodynamics for RNA secondary structure prediction. A Practical guide.” Zuker.m.athews, D.Turner, D.